10월 13일부터 16일까지 독일 하이델베르크에서 하는 The Non-Coding Genome 학회에 갑니다.
이히히. ^__^ 좋은 경험 있으시면 추천 바랍니다! (처음 가는 유럽이라 두근두근~)

© Junnn. CC-BY-NC-SA
혜식이의 열고 보는 세상
10월 13일부터 16일까지 독일 하이델베르크에서 하는 The Non-Coding Genome 학회에 갑니다.
이히히. ^__^ 좋은 경험 있으시면 추천 바랍니다! (처음 가는 유럽이라 두근두근~)
© Junnn. CC-BY-NC-SA
어느 학문 분야든 성숙하다보면 해당 분야의 역사와 철학에 대한 연구가 따라오게 된다.
학문이 생기게 된 배경과 발전 과정, 패러다임의 변화, 다른 학문에 대한 영향, 연구자들의 분야
고유적인 연구 방법을 관찰하는 것은 재미있지 않을 수가 없다.
최근에 PLoS Computational Biology에 생물정보학의 뿌리라는
기사가 올라왔다.
유전체 모델이나 RNA 2차구조 같은 것을 촘스키식 문법으로 다룬 것으로 유명한
David Searls가 쓴 생물정보학의 역사에 대한 글인데, 깊게 잘 다루었다.
철학적인 생각은 글에 남겨두고, 의외로 모르고 지나가기엔 너무 아쉬웠을 만한 재미있는 사실 몇 가지만 추려보면,
보통 어디서 트렌드따라 뚝 떨어진 신생융합듣보잡 취급을 많이 받는 생물정보학이지만 의외로 뿌리는 깊다. +_+
small RNA 시퀀싱에서는 리드보다 RNA가 더 짧아서, 5′ 끝부터 읽을 경우에는 3′ 어댑터 시퀀스가 나오고, 프라이머를 뒤집어서 3′ 끝부터 읽으면 5′ 시퀀스가 나올 수 밖에 없다. small RNA가 아니더라도 CLIP에서는 보통 바인딩 사이트를 정확히 알기위해 짧게 쳐내서 시퀀싱하는 경향이 있어서, 보통 30nt 안쪽으로 들어오는 편이라 시퀀싱한 뒤에 어댑터 제거가 꼭 필요하다.
|
1
2
|
AACTGTTTGCAGAGGAAACTGAATCTCGTATGCCGT – hsa–miR–452 뒤에 Illumina SRA 1.5 3‘ 어댑터
<—– miR-452 ——><– 3′ adapter
|
그렇지만 역시나 PCR 오류, 시퀀싱 오류, 어댑터 불량 등등 수많은 잡음때문에 역시 단순 문자열 비교로는 잘 안 통한다. 그래서 정규식을 쓰기도 하는데 영 속도가 만족스럽지 못하고, 모든 자리에서 어댑터 시퀀스랑 비교해서 미스매치를 세는 등의 방법(HTSeq 패키지)을 쓰기도 하는데, 갭을 전혀 허용하지 않아서 어댑터 합성 품질이 안 좋은 경우는 놓치는 것이 너무 많아서 결과를 보면 답답~하다.
최근 많이 쓰이는 방법으로 Needleman-Wunsch와 Smith-Waterman을 섞어서 어댑터의 5′ 끝에게는 지역정렬처럼 아무데서나 시작하게 하고, 3′ 끝에는 전체정렬처럼 끝까지 가게 하는 것이 있다. 그런데, 소프트웨어는 홈페이지에 오거나, 저자에게 말하면 준다고 다들 써 놓고서는 정작 홈페이지에 가면 아무 것도 없고, 메일 보내면 묵묵부답이라, 1년 넘게 정규식으로 불쌍하게 쓰다가, 결국 큰 마음먹고 토요일 밤을 투자했다. +_+
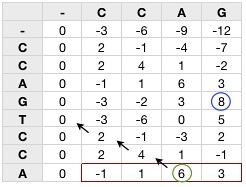
실제로는 affine gap penalty를 써서 행렬이 3개지만, 그냥 linear gap penalty를 쓴 경우라고 하고 행렬을 예를 들어 보면,
 Watch movie online The Transporter Refueled (2015)
Watch movie online The Transporter Refueled (2015)
이렇게 하면 짠~