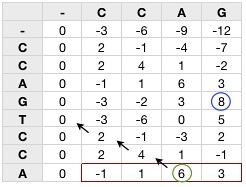
<span class=“kn”>import</span> <span class=“nn”>ctypes</span>
<span class=“n”>cranksum</span> <span class=“o”>=</span> <span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>cdll</span><span class=“o”>.</span><span class=“n”>LoadLibrary</span><span class=“p”>(</span><span class=“s”>'./cranksum.so'</span><span class=”p”>)</span>
<span class=“n”>crs</span> <span class=“o”>=</span> <span class=“n”>cranksum</span><span class=“o”>.</span><span class=“n”>clustered_rank_sum</span>
<span class=“n”>crs</span><span class=“o”>.</span><span class=“n”>restype</span> <span class=“o”>=</span> <span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>POINTER</span><span class=“p”>(</span><span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>c_double</span> <span class=“o”>*</span> <span class=“mf”>4</span><span class=“p”>)</span>
<span class=“n”>freecrs</span> <span class=“o”>=</span> <span class=“n”>cranksum</span><span class=“o”>.</span><span class=“n”>free_clustered_rank_sum_result</span>
<span class=“k”>def</span> <span class=“nf”>clustered_rank_sum</span><span class=“p”>(</span><span class=“n”>X</span><span class=“p”>,</span> <span class=“n”>grp</span><span class=“p”>,</span> <span class=“n”>cluster</span><span class=“p”>):</span>
<span class=“n”>N</span> <span class=“o”>=</span> <span class=“nb”>len</span><span class=“p”>(</span><span class=“n”>X</span><span class=“p”>)</span>
<span class=“n”>ret</span> <span class=“o”>=</span> <span class=“n”>crs</span><span class=“p”>((</span><span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>c_double</span> <span class=“o”>*</span> <span class=“n”>N</span><span class=“p”>)(</span><span class=“o”>*</span><span class=“n”>X</span><span class=“p”>),</span> <span class=“p”>(</span><span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>c_int</span> <span class=“o”>*</span> <span class=“n”>N</span><span class=“p”>)(</span><span class=“o”>*</span><span class=“n”>grp</span><span class=“p”>),</span>
<span class=“p”>(</span><span class=“n”>ctypes</span><span class=“o”>.</span><span class=“n”>c_int</span> <span class=“o”>*</span> <span class=“n”>N</span><span class=“p”>)(</span><span class=“o”>*</span><span class=“n”>cluster</span><span class=“p”>),</span> <span class=“n”>N</span><span class=“p”>)</span><span class=“o”>.</span><span class=“n”>contents</span>
<span class=“n”>r</span> <span class=“o”>=</span> <span class=“p”>{</span><span class=“s”>'S'</span><span class=”p”>:</span> <span class=”n”>ret</span><span class=”p”>[</span><span class=”mf”>0</span><span class=”p”>],</span> <span class=”s”>'E.S'</span><span class=”p”>:</span> <span class=”n”>ret</span><span class=”p”>[</span><span class=”mf”>1</span><span class=”p”>],</span> <span class=”s”>'Var.S'</span><span class=”p”>:</span> <span class=”n”>ret</span><span class=”p”>[</span><span class=”mf”>2</span><span class=”p”>],</span> <span class=”s”>'z.stat'</span><span class=”p”>:</span> <span class=”n”>ret</span><span class=”p”>[</span><span class=”mf”>3</span><span class=”p”>]}</span>
<span class=“n”>freecrs</span><span class=“p”>(</span><span class=“n”>ret</span><span class=“p”>)</span>
<span class=“k”>return</span> <span class=“n”>r</span>
<span class=“n”>X</span> <span class=“o”>=</span> <span class=“p”>[</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>4</span><span class=“p”>,</span><span class=“mf”>2</span><span class=“p”>,</span><span class=“mf”>4</span><span class=“p”>,</span><span class=“mf”>6</span><span class=“p”>,</span><span class=“mf”>7</span><span class=“p”>,</span><span class=“mf”>4</span><span class=“p”>,</span><span class=“mf”>7</span><span class=“p”>,</span><span class=“mf”>8</span><span class=“p”>]</span>
<span class=“n”>grp</span> <span class=“o”>=</span> <span class=“p”>[</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>]</span>
<span class=“n”>cluster</span> <span class=“o”>=</span> <span class=“p”>[</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>1</span><span class=“p”>,</span><span class=“mf”>2</span><span class=“p”>,</span><span class=“mf”>2</span><span class=“p”>,</span><span class=“mf”>2</span><span class=“p”>,</span><span class=“mf”>2</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>,</span><span class=“mf”>0</span><span class=“p”>]</span>
<span class=“n”>print</span> <span class=“n”>clustered_rank_sum</span><span class=“p”>(</span><span class=“n”>X</span><span class=“p”>,</span> <span class=“n”>grp</span><span class=“p”>,</span> <span class=“n”>cluster</span><span class=“p”>)</span>