small RNA 시퀀싱에서는 리드보다 RNA가 더 짧아서, 5′ 끝부터 읽을 경우에는 3′ 어댑터 시퀀스가 나오고, 프라이머를 뒤집어서 3′ 끝부터 읽으면 5′ 시퀀스가 나올 수 밖에 없다. small RNA가 아니더라도 CLIP에서는 보통 바인딩 사이트를 정확히 알기위해 짧게 쳐내서 시퀀싱하는 경향이 있어서, 보통 30nt 안쪽으로 들어오는 편이라 시퀀싱한 뒤에 어댑터 제거가 꼭 필요하다.
|
1
2
|
AACTGTTTGCAGAGGAAACTGAATCTCGTATGCCGT – hsa–miR–452 뒤에 Illumina SRA 1.5 3‘ 어댑터
<—– miR-452 ——><– 3′ adapter
|
그렇지만 역시나 PCR 오류, 시퀀싱 오류, 어댑터 불량 등등 수많은 잡음때문에 역시 단순 문자열 비교로는 잘 안 통한다. 그래서 정규식을 쓰기도 하는데 영 속도가 만족스럽지 못하고, 모든 자리에서 어댑터 시퀀스랑 비교해서 미스매치를 세는 등의 방법(HTSeq 패키지)을 쓰기도 하는데, 갭을 전혀 허용하지 않아서 어댑터 합성 품질이 안 좋은 경우는 놓치는 것이 너무 많아서 결과를 보면 답답~하다.
최근 많이 쓰이는 방법으로 Needleman-Wunsch와 Smith-Waterman을 섞어서 어댑터의 5′ 끝에게는 지역정렬처럼 아무데서나 시작하게 하고, 3′ 끝에는 전체정렬처럼 끝까지 가게 하는 것이 있다. 그런데, 소프트웨어는 홈페이지에 오거나, 저자에게 말하면 준다고 다들 써 놓고서는 정작 홈페이지에 가면 아무 것도 없고, 메일 보내면 묵묵부답이라, 1년 넘게 정규식으로 불쌍하게 쓰다가, 결국 큰 마음먹고 토요일 밤을 투자했다. +_+
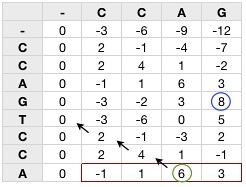
실제로는 affine gap penalty를 써서 행렬이 3개지만, 그냥 linear gap penalty를 쓴 경우라고 하고 행렬을 예를 들어 보면,
- 시퀀스 리드: CCAGTCCA
- 어댑터 시퀀스: CCAG
- 점수: match 2, mismatch -3, gap penalty -3
 Watch movie online The Transporter Refueled (2015)
Watch movie online The Transporter Refueled (2015)
이렇게 하면 짠~
이런 데이터 다루는구나~ 재미있겠다~^^
오오~~ biopython에 넣어주세요~
찬석: 크.. 맨날 머리를 굴리는 종류의 것을 하지는 않아.. ㅋㅋ;;; C가 이렇게 재미있는지 한참 안 해보니 알겠네 ^_^;;
yong27: 오… 일반화 하기엔 좀 번거로워서 -ㅇ-;;;;;